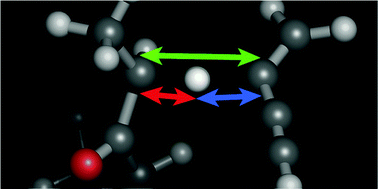
Bhoorasingh & West
PCCP 2015, 32173-32182
Complex Reactions

Machine Learning
The exhaustive exploration and characterization of the relevant elementary chemical pathways on the underlying potential energy surface is an extremely hard problem, considering the 3N–6 degrees of freedom of an N-atom chemical structure, combined with the expense of accurate calculations of the equilibrium geometries and energies of the molecular structures. The main goal of the work addressing these problems is to automate the discovery of fundamentally new reactions pathways and generalize them into reaction families both in gas-phase and heterogeneous catalyst systems. We are working on creating robust and efficient search algorithms and global strategies to find all reaction pathways by significantly improving current strategies and creating new ones by also invoking machine learning approaches.
KinBot and Sella
Background
Our KinBot code has efficient algorithms to find typical reactions for C, H, O, N, and S atom containing molecules. It demonstrated that it is possible to explore gas-phase chemical reactions relevant for combustion and atmospheric chemistry using the information about breaking and forming bonds and geometrical considerations in a heuristic framework. KinBot is currently coupled to Gaussian and Molpro. Our approach is efficient because the algorithm incorporates hard-won chemical knowledge, but it can only discover pathways in new systems that follow established mechanistic motifs, which are translated into the 3-D space of molecular structures by the code. Nevertheless, it is possible that a given motif has never been explored for a structure, and therefore KinBot can find, in this sense, new reactions. KinBot is able to propose many more saddle points than a human could, and discover unexpected or overlooked but important pathways, and in any case dramatically accelerates exploration for H, C, O and S-containing molecules in the context of gas-phase oxidation chemistry. The gas-phase core part of KinBot is being developed under the Combustion-Pele Exascale Computing Project (ECP), and grew out of an Early Career LDRD project at Sandia.
Developments under ECC
In ECC we are extending our fleet of PES searching codes. In KinBot we are adopting the current strategies to metal or metal oxide / gas phase catalytic systems. Our new code, Sella, is being developed within the ECC to locate and optimize first order saddle points on the potential energy surfaces of complex gas-phase and condensed-phase chemical systems. Whereas existing open source utilities for the optimization of saddle points typically require manually generated initial guesses, Sella will be able to perform automatic global searches for saddle points in complex chemical systems with a large number of reaction pathways using exascale computer resources. Furthermore, Sella will exhibit superior efficiency and stability for local saddle point optimization due to its sophisticated Hessian matrix approximation, which is used for both identification and optimization of saddle points. While we are targeting NWChem as the primary quantum chemistry code, Sella can be coupled with a number of different quantum chemistry packages through the Atomic Simulation Environment (ASE). Implementation of pathway searches begin with the currently available DFT and MP2 methods in NWChem, and will add random phase approximation methods for heterogeneous systems as we implement them. We also connect our codes to the Arrows app of NWChem. Our codes rely on underlying GPU-enabled (and Intel MIC) NWChem performance, but are also parallel at the high level.
References
AutoTST
Background
Often many reactions in a detailed mechanism have very similar saddle points, e.g. for hydrogen abstractions, and a de-novo unbiased pathway discovery tool is not required. Instead a highly efficient way of calculating up to thousands of reactions of the same type (but with different reactants) is more helpful. This is the aim of AutoTST, where we want to rapidly evaluate reaction properties (barrier heights, transition state structures, etc.) for many homologous reactions and cast the resulting information into mineable data.
Developments under ECC
We extend the AutoTST approach to reactions occurring on catalyst surfaces. The principles of predicting the interatomic distances at the saddle point remain, but use double-ended search such as the nudged elastic band method implemented using the ASE toolkit to facilitate interfacing with a variety of quantum codes and established geometry optimization methods. To accelerate the pathway optimization and saddle point search we use an artificial neural network representation of the PES, exploring the potential synergy with sparse representation tools.
Team
- Lead
- Judit Zádor (SNL)
- Senior researchers
- Habib N. Najm (SNL)
- Khachik Sargsyan (SNL)
- Richard H. West (Northeastern)
- Students and postdocs
- Eric D. Hermes (SNL)
- Maciej Gierada (SNL)
- Nathan Harms (Northeastern)
- David Farina (Northeastern)
References
N.D. Harms, C. Underkoffler, A.M. Payne, R.H. West . AutoTST: A framework to perform automated transition state theory calculations. Work in Progress Poster 1P003. 37th International Symposium on Combustion, Dublin, Ireland. 30 July 2018
